
|
|
QuartataWeb
Background
Data on protein-drug interactions are rapidly increasing and being collected in databases (DBs) such as DrugBank. These DBs usually contain information on known/observed interactions, while the lack of data for a given protein-drug pair does not necessarily mean that those protein-drug molecules are not interacting. Indeed, recent studies supported by both computations and experiments indicate that many drugs have side effects (i.e. they target multiple proteins) other than those known and compiled in DrugBank. Their promiscuity may be exploited for designing ‘repurposable’ drugs or developing polypharmacological treatments. Efficient identification of such potential interactions is an important goal that may be accelerated by machine learning methods. There is a need for efficient identification of such data, and efficient dissemination of results.
In this tutorial, we will describe the following:
1.2 The significance and purpose of QuartataWeb
1.3 The methodology used to build QuartataWeb
1.4 The protocol for usage
1.5 Chemical-target interaction prediction confidence scores and how to interpret them
1.6 Chemical-chemical and target-target similarity scores and how to interpret them
1.7 The interactive network visualization
1.8 An example use of QuartataWeb for drug repurposing
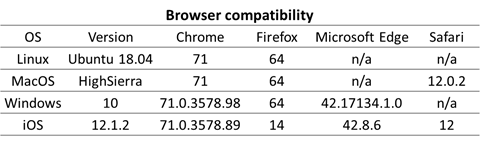
The website is compatible with all modern browsers (Chrome, Firefox, Microsoft Edge and Safari) in Windows, Linux, MacOS as well as the iOS.
Predicting CTIs and analyzing their effects on cellular pathways are of great importance in drug discovery and are necessary for the development of
QuartataWeb solves this problem by learning latent factor models (LFMs) to predict CTIs by applying probabilistic matrix factorization (PMF) method on known CTIs extracted from the DrugBank v5 and STITCH v5- the latest stable version currently available. The LFMs enable large scale CTI prediction, chemical-chemical similarity and target-target similarity prediction based on CTI patterns. To infer pathways and biological functions, QuartataWeb utilizes and statistically analyzes the human pathways and functional annotations in KEGG Pathways DB and GO Annotations DB, (GOA). Thus, for a given input set of chemicals (similar to chemogenomics screens), users can obtain consolidated pathways or enriched GO terms, through large ensembles of known and predicted targets.
To use the tool, please select one of the Data sources: DrugBank or STITCH. DrugBank is the default. FDA approved drugs, or all drugs in DrugBank, can be also selected in the Drug group. There are two input types can be selected: “Chemical and/or target” and “A list of chemicals, targets, or chemical combinations”.
To search for a drug in DrugBank, please input the DrugBank identifier (ID), or common drug name (such as "Sunitinib", "Atorvastatin", or "Aspirin"). The DrugBank IDs are of the form "DB12345" where the first two characters are "DB", and the remaining numbers identify the specific drug. Identifying the drug in this manner is guaranteed to work as the DrugBank ID uniquely selects the drug of interest. The DrugBank ID is the preferred input of drugs.
To search for a chemical when the STITCH data source is selected, please input the PubChem compound ID (CID) or common chemical name (such as “Risperidone”). PubChem CID is the preferred input of chemicals.
To search for a target, the user could enter the UniProt enter ID of the target. Alternatively, you can enter gene IDs from GenAtlas, or common name of a protein. Both the UniProt ID and gene ID of the targets are the preferred input.
Having entered the drug and/or target of interest; the user has three query choices:
For each DrugBank query, the user has the option to select "Secondary Interactions" when performing the query. If this option box is checked, then QuartataWeb displays the ‘second generation of neighbors’ in the network of interactions. The network is visualized by an interactive graph. Interactive visualization enables users to quickly see and select possible shared drugs/targets among the known and predicted interaction partners.
1.5 Chemical-Target Interaction Prediction
The user can perform one of the three following operations:
Interpreting Confidence Scores for Chemical-Target Interactions
The reported interaction confidence level, or score, has a broad range. To put these confidence scores into context the following guide can be used:
1.6 Chemical- Chemical Similarity and Target-Target Similarity Calculations
Chemical- Chemical Similarity
The user can perform the two following operations:
Target-Target Similarity Calculation
The operation mechanism is identical to the drug-drug similarity calculation described above, using target IDs instead of drug IDs.
Interpreting Chemical-Chemical and Target-Target Similarity Scores
We compute the chemical-chemical and target-target
similarities using cosine
similarity. Therefore the
values representative of the correlation between the queries pairs, range from
-1 (anti-correlated) to 1 (highly similar), the value of zero indicating lack
of correlation/similarity. The similarities between all chemical-chemical and
target-target pairs are distributed as shown in the histograms below.
When analyzing these similarity scores, the user can refer to the following guide:
1.7 Interactive Network Visualization
Whenever results are presented to the user, this is through an interactive network visualization interface. The interface facilitates the visual inspection of the results and has an overall view of the network on the spot. The node links represent the type of interaction between the linked nodes using the color coding: red for predicted interactions, grey for known interactions. The thickness of the edge indicates the confidence level of the interaction. The color of the nodes denotes whether the node represents a chemical (red) or target (blue). The network can be download as SVG image or JSON formats by clicking the "Download Network (SVG)" button or "Download Network (json)" link in the “Results” panel.
1.8 Example: Drug Repurposing against GBRT
In this section, we demonstrate the use of QuartataWeb by way of an example. Essentially, the concepts and methods described in the previous sections will be utilized within the framework of a concrete example to help the user better grasp the use of QuartataWeb.
We will set up the example as follows: suppose you would like to modulate the function of GBRT (GABA receptor subunit theta). This might be motivated by the fact that the protein has been implicated in a pathological process; or the need to interrogate the system of interest on how GBRT modulation might influence a pathway of interest, to give a few examples.
Without QuartataWeb, you would be forced to look at various DBs and find only a few known drug interactions for GBRT: DrugBank v5.1 lists 53 drug interactions. While with QuartataWeb, you simply go to the QuartataWeb main page, enter "GBRT" to the target box and click the "Submit" button. You can immediately view both the known drugs, as well as the top 20 drugs (with confidence scores > 90%) predicted to be the potential interaction partners of GBRT.
Based on this information, you can choose to look for other drugs that are most similar to Thiopental, for example. QuartataWeb can be used for this purpose as well. You can simply check the “chemical-chemical similarity” radio box button on the navigation bar and enter "Thiopental". On this page, there are two input boxes: one for the main chemical of interest (here is the drug "Thiopental"), and another for the optional second chemical to compute its similarity to the primary chemical. For chemical-chemical similarity queries, QuartataWeb calculates the cosine similarity of the LVs of the chemicals. If there is only one input, QuartataWeb reports the top n (20 by default, user adjustable) most similar chemical s. If you input two chemical s, QuartataWeb computes the similarity between these two chemical s and reports it along with the targets of these chemical s. Since the similarity is calculated using a LFM trained from CTIs, this chemical-chemical similarity is fundamentally different from those inferred from 2D or 3D structure-based comparisons. Returning to our example, Thiopental, (which is an anesthetic) has been found to have the highest similarity to Etomidate (another anesthetic). The second most similar chemical is Isoflurane (which is also an anesthetic). As highlighted by these examples, the LFM finds therapeutically similar chemicals and is therefore capable of providing researchers with new directions to pursue. The exact same functionality exists for targets and can be used by checking the “target-target similarity” radio box button.