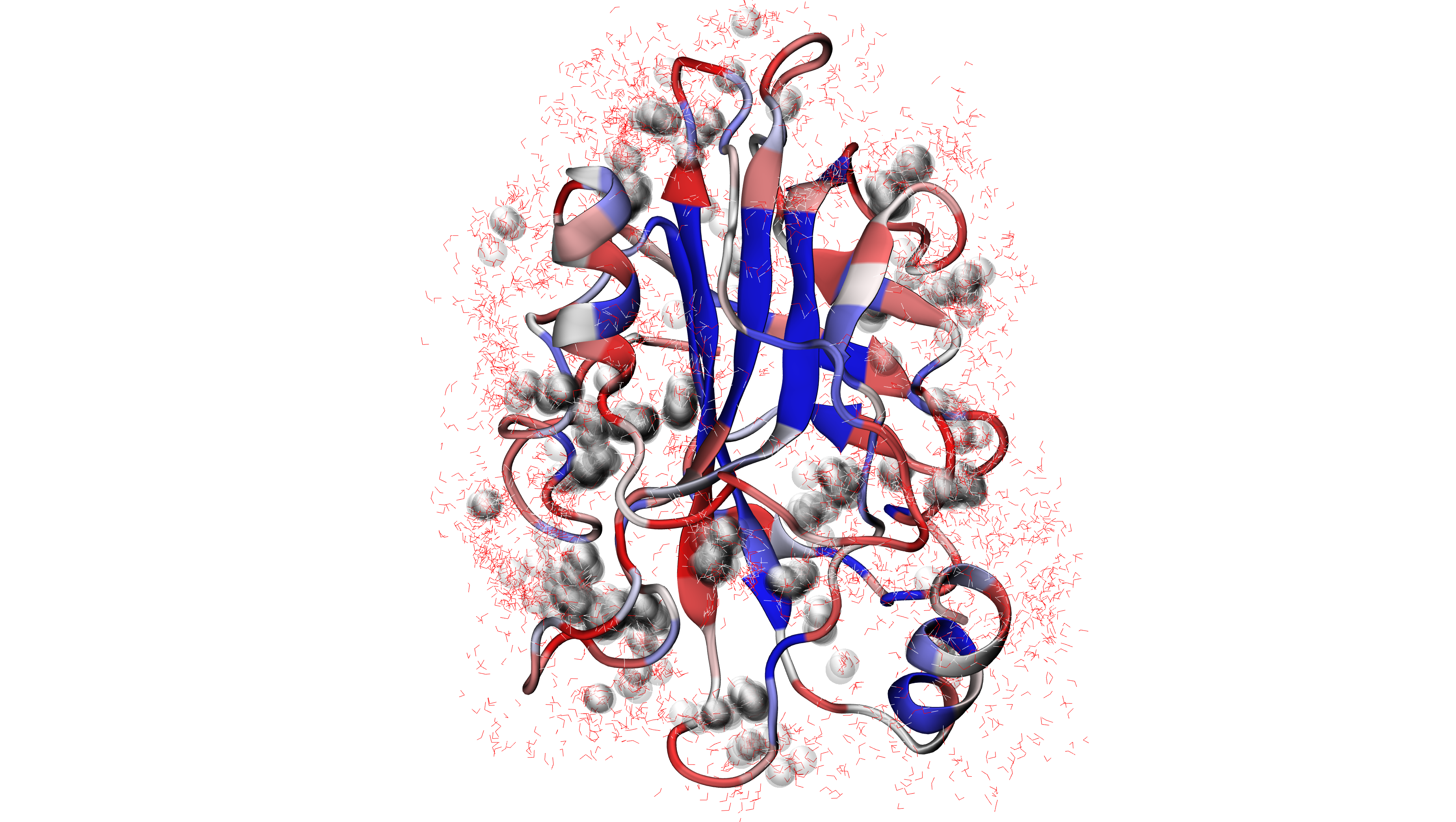
Water bridges detection in an Ensemble PDB¶
This time we will use an ensemble stored in a multi-model PDB, which contains 50 frames from MD simulations from PE-binding protein 1 (PDB: 1BEH). Simulations were performed using NAMD_ and saved as a multi-model PDB using VMD.
We need to remember to align the protein structure before performing the analysis.
Otherwise, when all structures are uploaded to the visualization program, they will
be spread out in space. We could do this inside ProDy by converting the Atomic
object to an Ensemble object and using its Ensemble.iterpose() method to do this,
but here we demonstrate the process for parsing a multi-model PDB file directly.
Initial analysis¶
In [1]: ens = 'pebp1_50frames.pdb'
In [2]: coords_ens = parsePDB(ens)
In [3]: bridgeFrames_ens = calcWaterBridgesTrajectory(coords_ens, coords_ens)
@> 20195 atoms and 51 coordinate set(s) were parsed in 1.88s.
@> Frame: 0
@> 161 water bridges detected.
@> Frame: 1
@> 127 water bridges detected.
@> Frame: 2
@> 168 water bridges detected.
@> Frame: 3
@> 132 water bridges detected.
@> Frame: 4
@> 142 water bridges detected.
@> Frame: 5
@> 166 water bridges detected.
@> Frame: 6
@> 150 water bridges detected.
@> Frame: 7
@> 159 water bridges detected.
@> Frame: 8
@> 147 water bridges detected.
@> Frame: 9
@> 136 water bridges detected.
@> Frame: 10
@> 127 water bridges detected.
@> Frame: 11
@> 135 water bridges detected.
...
...
Analysis of the results is similar to that presented in trajectory analysis.
Below are examples showing which residues are most frequently involved in water bridge
formation (calcBridgingResiduesHistogram()), details of those interactions
(calcWaterBridgesStatistics()), and results saved as a PDB structure for further
visualization (savePDBWaterBridgesTrajectory()). Other functions can be explored
in the trajectory analysis.
In [4]: calcBridgingResiduesHistogram(bridgeFrames_ens)
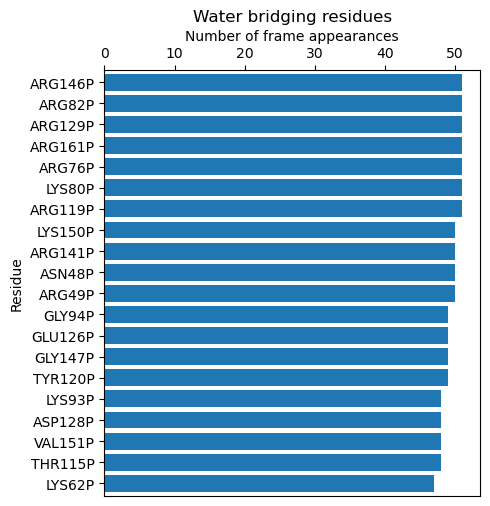
[('VAL34P', 1),
('VAL177P', 1),
('PRO43P', 1),
('LEU41P', 2),
('MET92P', 2),
('VAL164P', 3),
('LEU14P', 3),
('TYR169P', 3),
('PHE154P', 4),
.
.
('ARG49P', 50),
('ASN48P', 50),
('ARG141P', 50),
('LYS150P', 50),
('ARG119P', 51),
('LYS80P', 51),
('ARG76P', 51),
('ARG161P', 51),
('ARG129P', 51),
('ARG82P', 51),
('ARG146P', 51)]
In [5]: analysisAtomic_ens = calcWaterBridgesStatistics(bridgeFrames_ens,
...: coords_ens)
...:
In [6]: for item in analysisAtomic_ens.items():
...: print(item)
...:
@> RES1 RES2 PERC DIST_AVG DIST_STD
@> VAL3P HSE26P 19.608 5.581 0.696
@> ASP4P SER6P 13.725 3.817 0.560
@> SER6P LYS7P 43.137 4.394 1.114
@> LYS7P GLU36P 1.961 6.088 0.000
@> LYS7P LEU37P 7.843 6.353 0.433
@> GLY10P SER13P 43.137 4.759 0.612
@> GLY10P ARG76P 11.765 5.309 0.586
@> LEU12P SER13P 45.098 2.767 0.080
@> SER13P GLU16P 25.490 4.449 1.133
@> GLN15P ASP18P 7.843 3.732 0.174
@> GLU16P ARG82P 45.098 4.550 1.086
@> GLU16P VAL17P 17.647 3.438 0.952
@> GLU16P LYS150P 21.569 5.056 0.929
@> GLU16P GLU83P 9.804 5.476 1.138
@> GLU16P ALA152P 7.843 7.307 0.450
@> VAL17P GLU83P 1.961 7.262 0.000
@> VAL17P LYS150P 13.725 6.303 0.572
@> GLN22P GLU126P 33.333 6.458 1.216
@> GLN22P HSE23P 37.255 4.738 0.669
@> HSE23P GLU126P 7.843 7.911 0.239
@> PRO24P ASP56P 17.647 5.592 0.910
@> THR28P SER52P 43.137 3.970 0.677
@> THR28P ILE53P 5.882 5.849 0.027
@> TYR29P THR51P 7.843 3.583 0.286
@> ALA30P ARG49P 17.647 5.206 0.304
...
...
In [7]: savePDBWaterBridgesTrajectory(bridgeFrames_ens, coords_ens, ens[:-4]+'_ens.pdb')
@> All 51 coordinate sets are copied to pebp1_50frames Selection 'protein' + pebp1_50frames Selection 'same residue as...6074 4190 14360'.
@> All 51 coordinate sets are copied to pebp1_50frames Selection 'protein' + pebp1_50frames Selection 'same residue as...9718 17936 7184'.
@> All 51 coordinate sets are copied to pebp1_50frames Selection 'protein' + pebp1_50frames Selection 'same residue as...947 10043 11756'.
..
..
Detecting water centers¶
The previous function generated multiple PDB files in which we can find protein and
water molecules for each frame that form water bridges with the protein structure.
Now we can use another function findClusterCenters() which will extract
water centers (they refer to the oxygens from water molecules that are forming
clusters). We need to provide a file pattern as show below. Now all the PDB files
with prefix 'pebp1_50frames_ens_' will be analyzed.
In [8]: findClusterCenters('pebp1_50frames_ens_*.pdb')
@> 3269 atoms and 1 coordinate set(s) were parsed in 0.11s.
@> 3161 atoms and 1 coordinate set(s) were parsed in 0.05s.
@> 3173 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> 3173 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> 3218 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> 3251 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> 3215 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> 3230 atoms and 1 coordinate set(s) were parsed in 0.03s.
@> 3230 atoms and 1 coordinate set(s) were parsed in 0.04s.
@> 3224 atoms and 1 coordinate set(s) were parsed in 0.03s.
@> 3158 atoms and 1 coordinate set(s) were parsed in 0.03s.
..
..
@> Results are saved in clusters_pebp1_50frames_ens_.pdb.
This function generated one PDB file with water centers. We used default values,
such as distC (distance to other molecule) and numC (min number of molecules
in a cluster), but those values could be changed if the molecules are more
widely distributed or we would like to have more numerous clusters.
Moreover, this function can be applied to different types of molecules by using
the selection parameter. We can provide the whole molecule, and by
default, the center of mass will be used as a reference.
Saved PDB files using savePDBWaterBridgesTrajectory() in the previous
step can be upload to VMD or other program for visualization:
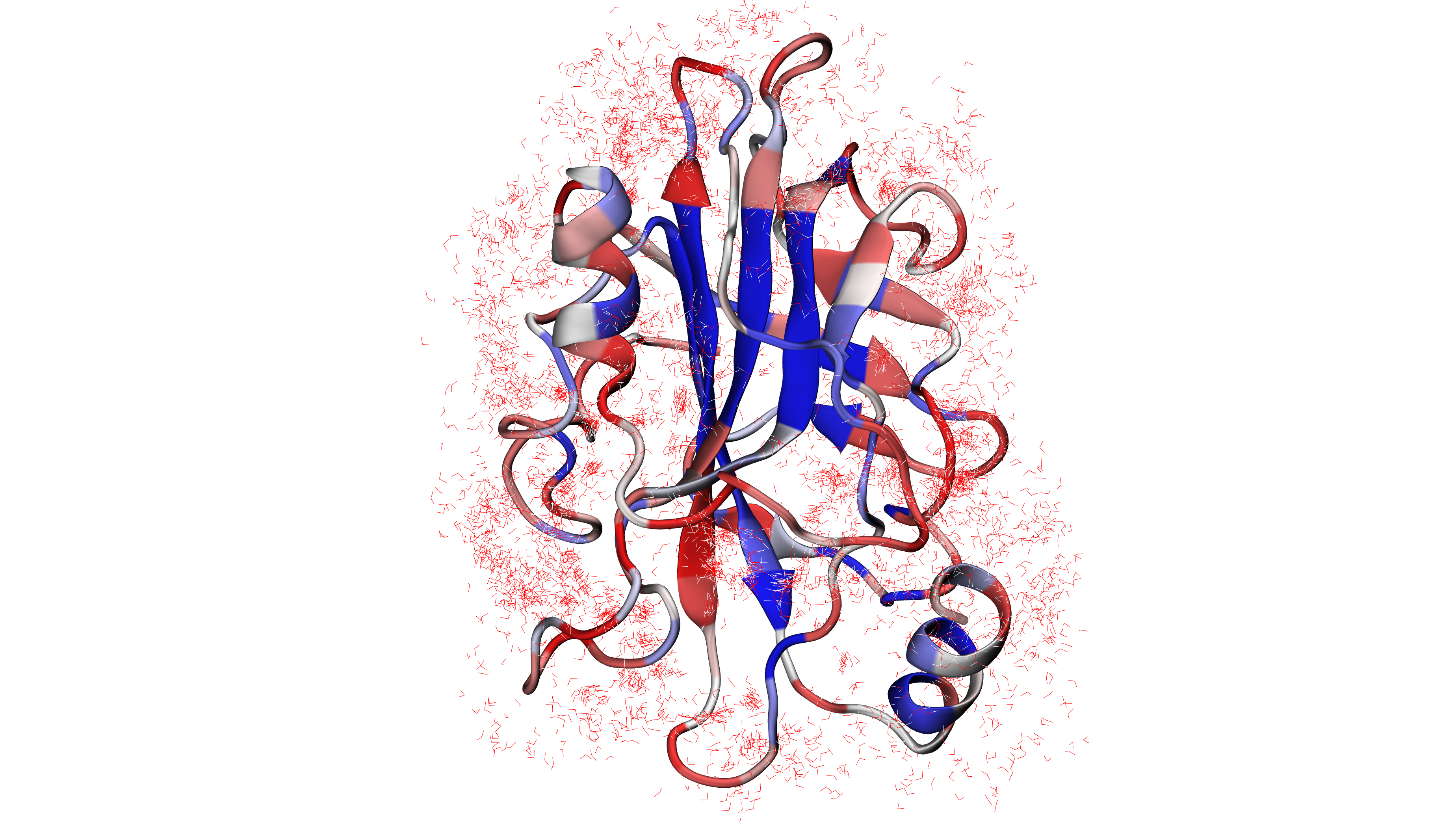
After uploading a new PDB file with water centers we can see the results as follows: