ProDy Basics¶
We start with importing everything from ProDy package:
In [1]: from prody import *
In [2]: from pylab import *
In [3]: ion()
Functions and classes are named such that they should not create a conflict with any other package. In this part we will familiarize with different categories of functions and methods.
File Parsers¶
Let’s start with parsing a protein structure and then keep working on that
in this part. File parser function names are prefixed with parse.
You can get a list of parser functions by pressing TAB after typing
in parse:
In [4]: parse<TAB>
parseArray parseCIFStream parseEMD parseHiC parseMSA
parsePDBHeader parsePQR parseSTAR parseChainsList parseDCD
parseEMDStream parseHiCStream parseNMD parsePDBStream parsePSF
parseSTRIDE parseCIF parseDSSP parseHeatmap parseModes
parsePDB parsePfamPDBs parseSparseMatrix
When using parsePDB(), usually an identifier will be sufficient,
If corresponding file is found in the current working directory, it will be
used, otherwise it will be downloaded from PDB servers.
Let’s parse structure 5uoj of p38 MAP kinase (MAPK):
In [5]: p38 = parsePDB('5uoj') # returns an AtomGroup object
In [6]: p38 # typing in variable name will give some information
Out[6]: <AtomGroup: 5uoj (3138 atoms)>
We see that this structure contains 2962 atoms.
Now, similar to listing parser function names, we can use tab completion to
inspect the p38 object:
In [7]: p38.num<TAB>
p38.numAtoms p38.numChains p38.numFragments p38.numSegments
p38.numBonds p38.numCoordsets p38.numResidues
This action printed a list of methods with num prefix. Let’s use some of them to get information on the structure:
In [8]: p38.numAtoms()
Out[8]: 3138
In [9]: p38.numCoordsets() # returns number of models
Out[9]: 1
In [10]: p38.numResidues() # water molecules also count as residues
Out[10]: 718
Analysis Functions¶
Similar to parsers, analysis function names start with calc:
In [11]: calc<TAB>
calcADPAxes calcChainsNormDistFluct calcCrossProjection
calcDistFlucts calcFractVariance calcADPs
calcCollectivity calcCumulOverlap calcENM
calcGNM calcAngle calcCovariance
calcDeformVector calcEnsembleENMs calcGyradius
calcANM calcCovOverlap calcDihedral
calcEnsembleSpectralOverlaps calcMeff calcCenter
calcCrossCorr calcDistance calcEntropyTransfer
calcMSAOccupancy calcMSF calcPairDeformationDist
calcPsi calcSignatureCollectivity calcSpecDimension
calcOccupancies calcPercentIdentities calcRankorder
calcSignatureCrossCorr calcSpectralOverlap calcOmega
calcPerturbResponse calcRMSD calcSignatureFractVariance
calcSqFlucts calcOverallNetEntropyTransfer calcPhi
calcRMSF calcSignatureOverlaps calcSubspaceOverlap
calcOverlap calcProjection calcShannonEntropy
calcSignatureSqFlucts calcTempFactors calcTransformation
calcTree
Let’s read documentation of calcGyradius() function and use it to
calculate the radius of gyration of p38 MAPK structure:
Plotting Functions¶
Likewise, plotting function names have show prefix and here is a list of them:
In [12]: show<TAB>
showAlignment showCrossProjection showDomainBar
showHeatmap showMeanMechStiff showNormDistFunct
showAtomicLines showCumulFractVars showDomains
showLines showMechStiff showNormedSqFlucts
showAtomicMatrix showCumulOverlap showEllipsoid
showLinkage showMode showOccupancies
showContactMap showDiffMatrix showEmbedding
showMap showMSAOccupancy showOverlap
showCrossCorr showDirectInfoMatrix showFractVars
showMatrix showMutinfoMatrix showOverlaps
showOverlapTable showScaledSqFlucts showSignatureCollectivity
showSignatureSqFlucts showVarianceBar showPairDeformationDist
showSCAMatrix showSignatureCrossCorr showSignatureVariances
showPerturbResponse showShannonEntropy showSignatureDistribution
showSqFlucts showProjection showSignature1D
showSignatureMode showTree showProtein
showSignatureAtomicLines showSignatureOverlaps showTree_networkx
We can use showProtein() function to make a quick plot of p38 structure:
In [13]: showProtein(p38);
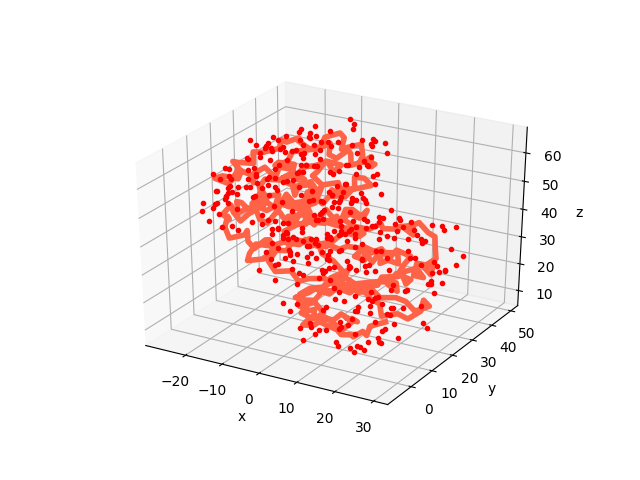
This of course does not compare to any visualization software that you might be familiar with, but it comes handy to see what you are dealing with.
Protein Structures¶
Protein structures (.pdb or .cif files) will be the standard input for most
ProDy calculations, so it is good to familiarize with ways to access and
manage PDB file resources.
Fetching PDB files¶
First of all, ProDy downloads PDB files when needed (these are compressed on the PDB webserver).
If you prefer saving decompressed files, you can use fetchPDB() function as
follows:
In [14]: fetchPDB('5uoj', compressed=False)
Out[14]: '5uoj.pdb'
Note that ProDy functions that fetch files or output files return filename upon successful completion of the task. You can use this behavior to shorten the code you need to write, e.g.:
In [15]: parsePDB(fetchPDB('5uoj', compressed=False)) # same as p38 parsed above
Out[15]: <AtomGroup: 5uoj (3138 atoms)>
We downloaded and save an uncompressed PDB file, and parsed it immediately.
PDB file resources¶
Secondly, ProDy can manage local mirrors of the PDB server or a local PDB folder, as well as using a server close to your physical location for downloads:
- One of the wwPDB FTP servers in US, Europe or Japan can be picked for downloads using
wwPDBServer().- A local PDB mirror can be set for faster access to files using
pathPDBMirror().- A local folder can be set for storing downloaded files for future access using
pathPDBFolder().
If you are in the Americas now, you can choose the PDB server in the US as follows:
In [16]: wwPDBServer('us')
If you would like to have a central folder, such as ~/Downloads/pdb,
for storing downloaded PDB files (you will need to make it), do as follows:
In [17]: mkdir ~/Downloads/pdb;
In [18]: pathPDBFolder('~/Downloads/pdb')
Note that when these functions are used, ProDy will save your settings
in .prodyrc file stored in your home folder.
Atom Groups¶
As you might have noticed, parsePDB() function returns structure data
as an AtomGroup object. Let’s see for p38 variable from above:
In [19]: p38
Out[19]: <AtomGroup: 5uoj (3138 atoms)>
You can also parse a list of .pdb files into a list of AtomGroup
objects:
In [20]: ags = parsePDB('5uoj', '3h5v')
In [21]: ags
Out[21]: [<AtomGroup: 5uoj (3138 atoms)>, <AtomGroup: 3h5v (9392 atoms)>]
If you want to provide a list object you need to provide an asterisk (*) to
let Python know this is a set of input arguments:
In [22]: pdb_ids = ['5uoj', '3h5v']
In [23]: ags = parsePDB(pdb_ids)
In [24]: ags
Out[24]: [<AtomGroup: 5uoj (3138 atoms)>, <AtomGroup: 3h5v (9392 atoms)>]
Data from this object can be retrieved using get methods. For example:
In [25]: p38.getResnames()
Out[25]: array(['ARG', 'ARG', 'ARG', ..., 'HOH', 'HOH', 'HOH'], dtype='|S6')
In [26]: p38.getCoords()
Out[26]:
array([[ 25.325, 3.794, 22.831],
[ 24.258, 4.528, 22.091],
[ 23.399, 3.547, 21.279],
...,
[ -1.774, 28.702, 32.891],
[ -1.648, 31.544, 31.756],
[-25.711, 35.476, 55.232]])
To get a list of all methods use tab completion, i.e. p38.<TAB>.
We will learn more about atom groups in the following chapters.
Indexing¶
An individual Atom can be accessed by indexing AtomGroup
objects:
In [27]: atom = p38[0]
In [28]: atom
Out[28]: <Atom: N from 5uoj (index 0)>
Note that all get/set functions defined for AtomGroup
instances are also defined for Atom instances, using singular
form of the function name.
In [29]: atom.getResname()
Out[29]: 'ARG'
Slicing¶
It is also possible to get a slice of an AtomGroup. For example,
we can get every other atom as follows:
In [30]: p38[::2]
Out[30]: <Selection: 'index 0:3138:2' from 5uoj (1569 atoms)>
Or, we can get the first 10 atoms, as follows:
In [31]: p38[:10]
Out[31]: <Selection: 'index 0:10:1' from 5uoj (10 atoms)>
Hierarchical view¶
You can also access specific chains or residues in an atom group. Indexing
by a single letter identifier will return a Chain instance:
In [32]: p38['A']
Out[32]: <Chain: A from 5uoj (718 residues, 3138 atoms)>
Indexing atom group with a chain identifier and a residue number will return
Residue instance:
In [33]: p38['A', 100]
Out[33]: <Residue: ASN 100 from Chain A from 5uoj (8 atoms)>
See Atomic classes for details of indexing atom groups and Hierarchical Views for more on hierarchical views.
ProDy Verbosity¶
Finally, you might have noticed that ProDy prints some information to the console after parsing a file or doing some calculations. For example, PDB parser will print what was parsed and how long it took to the screen:
@> 5uoj (./5uoj.pdb.gz) is found in the target directory.
@> 2962 atoms and 1 coordinate sets were parsed in 0.08s.
This behavior is useful in interactive sessions, but may be problematic for
automated tasks as the messages are printed to stderr. The level of verbosity
can be controlled using confProDy() function, and calling it as
confProDy(verbosity='none') will stop all information messages permanently.