Sample Conformations¶
In this part, we will sample conformations along ANM modes.
In [1]: from prody import *
In [2]: from pylab import *
In [3]: ion()
Load results¶
First, we load results produced in the previous part. If you are in the same Python session, you don’t need to do this.
In [4]: p38 = loadAtoms('p38.ag.npz')
In [5]: p38_anm = loadModel('p38_ca.anm.npz')
In [6]: p38_anm_ext = loadModel('p38_ext.nma.npz')
Sampling¶
We will use the sampleModes() function:
In [7]: ens = sampleModes(p38_anm_ext, atoms=p38.protein, n_confs=40, rmsd=1.0)
In [8]: ens
Out[8]: <Ensemble: Conformations along NMA Extended ANM p38 ca (40 conformations; 2834 atoms)>
This will produce 40 (n_confs) conformations with have an
average RMSD of 1.0 Å from the input structure.
We can write this ensemble in a .dcd for visualization in VMD:
In [9]: writeDCD('p38all.dcd', ens)
Analysis¶
Let’s analyze the Ensemble by plotting the RMSDs of all conformations
relative to the input structure:
In [10]: rmsd = ens.getRMSDs()
In [11]: hist(rmsd, density=False);
In [12]: xlabel('RMSD');
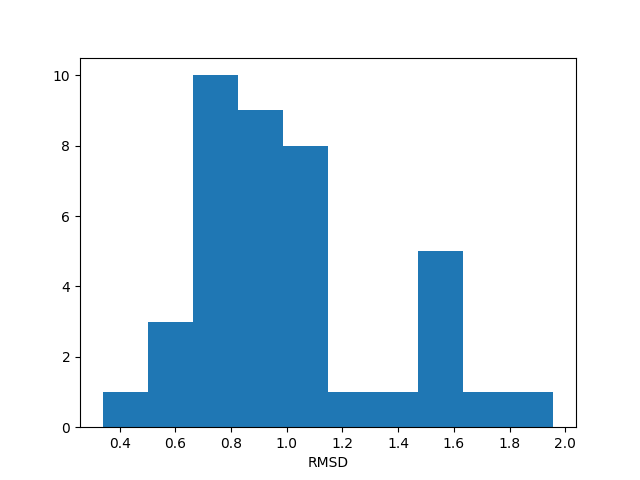
This histogram might look like a flat distribution due to the small size of the ensemble. For larger numbers of conformations it will get closer to a normal distribution.
Let’s see the projection of these conformations in the ANM slow mode space:
In [13]: showProjection(ens, p38_anm_ext[:3], rmsd=True);
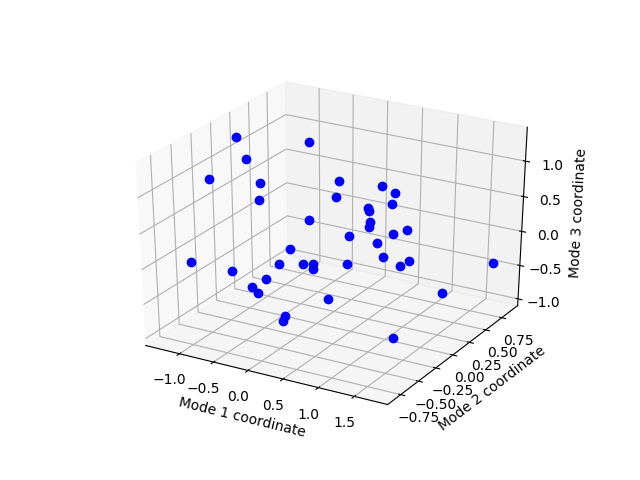
Write conformations¶
We will write them in p38_ensemble folder:
In [14]: mkdir -p p38_ensemble
Let’s add the conformations to the AtomGroup object and set
beta values of Cα atoms to 1 and of other atoms to 0:
In [15]: p38.addCoordset(ens.getCoordsets())
In [16]: p38
Out[16]: <AtomGroup: p38 (2834 atoms; active #0 of 41 coordsets)>
In [17]: p38.all.setBetas(0)
In [18]: p38.ca.setBetas(1)
In the next step, we will optimise the atom positions with a harmonic constraint on atoms with beta values of 1. The optimization aims to refine covalent geometry of atoms.We do not want the new Cα to change much to keep the refined ensemble diverse. We can easily verify that only Cα atoms have beta values set to 1:
In [19]: p38.ca == p38.beta_1
Out[19]: True
Now we write these conformations out:
In [20]: import os
In [21]: for i in range(1, p38.numCoordsets()): # skipping 0th coordinate set
....: fn = os.path.join('p38_ensemble', 'p38_' + str(i) + '.pdb')
....: writePDB(fn, p38, csets=i)
....:
Visualization¶
You can visualize all of these conformations using VMD as follows:
$ vmd -m p38_ensemble/*pdb