Abstract
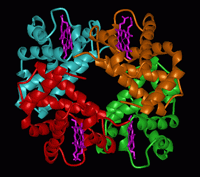
Coordinated motions of proteins are known to be relevant to their function. So far the study of protein dynamics largely relies on spectroscopic studies of part of the proteins or indirectly by comparing the conformations assumed in the presence of different substrates or ligands. While molecular simulations can also provide information on protein dynamics, these usually suffer from incomplete sampling of conformational space, and become prohibitively expensive when exploring the collective dynamics of large macromolecular structures. In this study, we explore the dynamics of a well-studied allosteric protein, hemoglobin (Hb), to show that a simple mechanical model based on Gaussian fluctuations of inter-residue distances can efficiently predict the collective dynamics of the tetramer in the liganded and unliganded forms; and more importantly the conformational transition between the relaxed (R) and tense (T) forms of Hb assumed in the unliganded and liganded conformations exactly coincides with the structural change driven by the global mode. This is the first demonstration of the intrinsic tendency of the Hb architecture to undergo the T ßà R transition, simply by the action of the most cooperative mechanical mode that is uniquely defined for the particular tetrameric fold.
|