
|
|
|
Drug Discovery |
|
|
Our lab utilizes quantitative systems pharmacology (QSP)
and computational modeling methods, structure-based
docking analyses, druggability simulations, pharacophore
modeling, and virtual screening to elucidate the mechanisms
of protein-drug interactions at the molecular and cellular
systems level, and help discover new drugs.
There has been a surge in recent years in the number of
QSP studies that exploit existing knowledge of protein-drug and
protein-ligand interactions. QSP approaches help reduce
wet lab work, assist in selecting lead compounds, in
assessing side effects and identifying repurposable drugs
(1-2).
We have developed
BalestraWeb
(1-2) for identifying repurposable drugs, and the
druggability suite
DruGUI
(3) for efficient evaluation of potential binding sites
and affinities on target proteins.
|
|
|
Overview of druggability simulation method
|
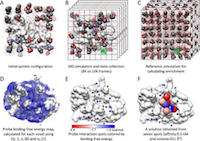
|
|
References:
1. Cobanoglu MC, Oltvai ZN, Taylor DL, Bahar I. (2014) BalestraWeb: Efficient, online evaluation of drug-target interactions Bioinformatics 31:131-3.
2. Cobanoglu MC, Liu C, Hu F, Oltvai ZN, Bahar I. (2013) Predicting drug-target interactions using probabilistic matrix factorization. J Chem Inf Model 53:3399-409.
3. Bakan A, Nevins N, Lakdawala AS, Bahar I (2012) Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules J Chem Theory Comput 8:2435-2447.
|
| |
| |
| |
|
|
| |
|

